单细胞测序的文章发了辣么多,我也想去凑凑热闹,怎么能轻松入手呢?
今天小编通过解读一篇发表在Cell Rep上的文章,来给大家讲一下八分的单细胞测序文章都做了哪些事?流程是什么样的?助你轻松上手单细胞测序!
- 文章标题:Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma
- 发表期刊:Cell REP
- 发表时间:October 31, 2017
- 影响因子:8.032
- GBM肿瘤细胞的巨大基因组和转录组异质性
- 肿瘤周围组织中单个GBM浸润细胞的表征
- 脑胶质瘤是最常见的原发性恶性脑癌,因其广泛的扩散性、复发性十分难以治疗。除了一些激进的治疗手段,如手术切除、放疗、化疗等,病人的存活率通常在12—18个月之间。
- 为了开发出更好的治疗方案,必须对肿瘤细胞(尤其是造成复发的浸润细胞)的分子层面进行更深入的研究。传统RNA测序虽然能够区分出脑胶质瘤的亚型,但无法分析肿瘤的异质性。
- 为了更好地掌握脑胶质瘤的扩散过程,作者运用单细胞RNA测序对4名病人的3,589个癌与癌旁细胞进行测序分析,成功区分出了由癌组织中心向癌周扩散的浸润性癌细胞。
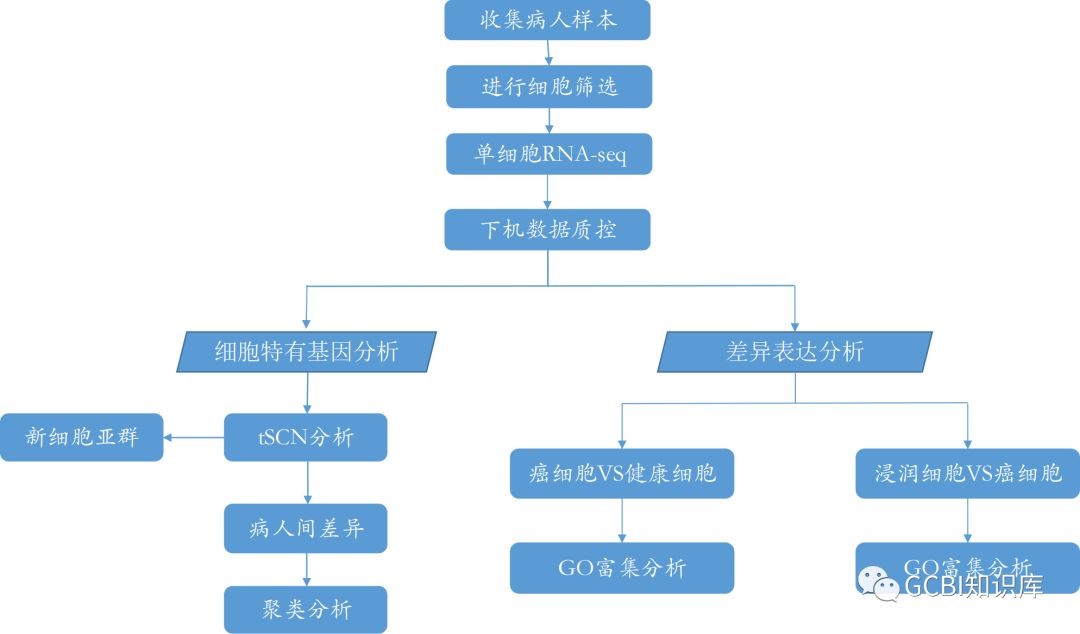
作者在本次实验中进行了转录组、基因组、免疫应答等多方面的分析,其中最值得关注的是单细胞RNA-seq的转录组分析。我们将以此为对象,对其在肿瘤方面的单细胞RNA-seq的研究思路进行参考和学习。
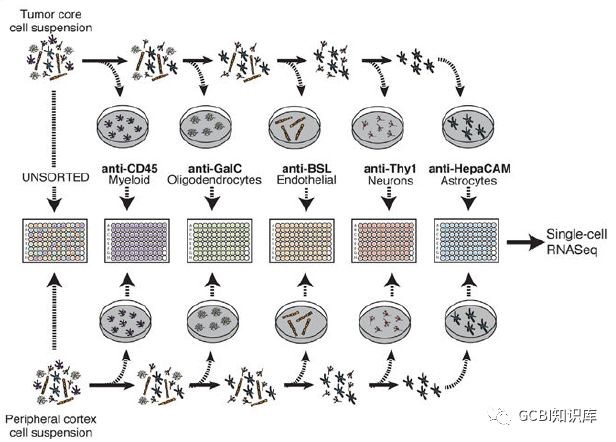
为了研究癌细胞的扩散机制,作者首先把细胞区分为癌细胞与癌周细胞两种。在实验初期,作者又发现在区分癌细胞亚群的时候,一定会有一部分非肿瘤细胞的存在,其中包括神经、血管和免疫细胞等。为了更好地利用单细胞测序技术把这些在普通RNA测序中含糊不清的部分清楚地剖析开来,作者利用特殊标志物对样本进行分选,将细胞人为地归类成不同的组别。最后对所有细胞进行单细胞RNA-seq。通过事先将细胞进行一次分类,能够更好地明确单细胞RNA-seq测序的后续分析流程,也能够提升对样本的把控程度。
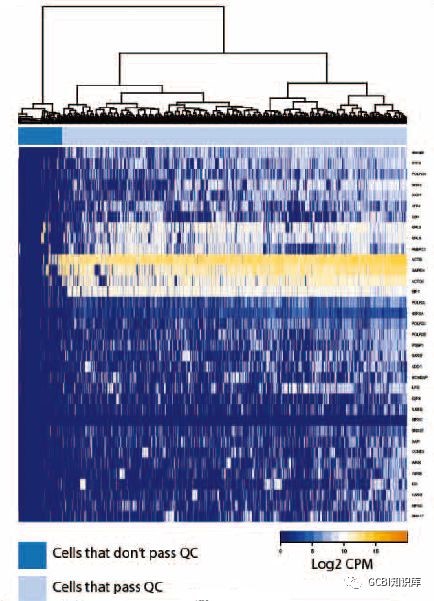
1.单细胞测序数据一次下机那么多,到手后第一步应该做什么?首先,为了对单细胞测序的下机数据进行质检,作者团队挑选了500个管家基因作为参考对象,将基因高表达的细胞数据视作有效数据。据此标准做出的细胞差异矩阵图对细胞进行聚类,直观地展示出数据质检结果。将所有细胞分为了3589个可用细胞,和456个不可用细胞。得到了质检结果后,就可以对区分出来的有效数据进行后续分析了。
2.QC后先做什么?细胞分型是第一步为了将所有细胞进行分类,作者运用了tSNE算法,通过鉴定不同细胞种类所特有的高表达基因,最终得到了12种细胞类型。那么问题就来了,如何鉴定出细胞特有的高表达基因呢?
为了达到这个目的,从而对细胞进行分类,作者使用了以下方法:首先,运用Wilcoxon 秩和检验,将每一簇细胞与其它所有细胞之间的基因表达关系进行检验。然后,查询现有的文献数据以及数据库(CNS)中的可靠数据,与实验结果进行交叉比对。最终,通过这些手段确定了用于鉴定细胞类型的特有高表达基因。
3.癌细胞特征分析(肿瘤测序必备流程)为了找到在癌细胞中表达差异最显著的基因,作者首先运用了DESeq2将癌细胞组与非癌细胞组这两种样本进行差异分析,观察癌细胞与非癌细胞之间的基因表达差异。分析结果得到了5143个差异基因,其中有3985个上调基因,和1158个下调基因。然后,作者添加了新的筛选条件,挑选出那些既在60%以上癌细胞表达,同时又只在20%以下非癌细胞中有表达的基因。
最终,得到了30个满足条件的基因。结果如图所示,不但直观地展现了在癌细胞高表达的基因,而且验证了其它学者之前所作出的研究结果。
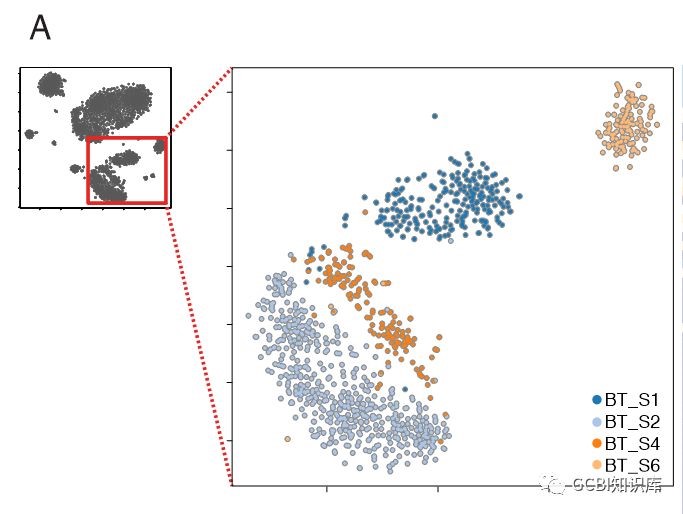
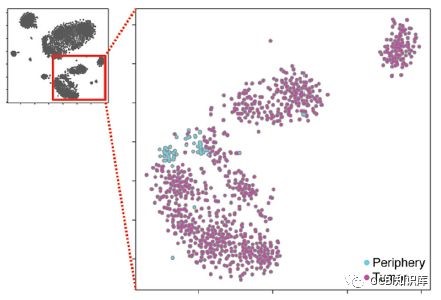
4. 至此,完成了肿瘤RNA测序的常用流程。但作为一个有追求的作者,必须充分发挥单细胞RNA-seq的作用。首先就是进一步地对癌细胞的异质性进行研究。作者对tSNE图中的癌细胞组进行放大,希望观察到不同病人间癌细胞差异。结果发现,4个病人的癌细胞最终确实被细分成了4个不同类群。
既然发现了这个现象,就要对其原因进行分析。为了深入挖掘造成异质性的原因,作者运用了Verhaak在其文章中 (Verhaak et al., 2010)使用的GMB亚型分类方法,希望发现病人间癌细胞的异质性是由于他们的癌细胞分属于不同的亚型而造成。但并没有得到预期的结果,癌细胞大多属于同一亚型,没有显著差异。于是作者对所有癌细胞的基因表达情况进行了聚类分析,发现不同病人癌细胞之间,从根本上不同的基因表达情况,是造成差异的主要因素。
5.癌细胞转移机制,本实验的终极研究目标。进行了一系列常规的QC、聚类、验证等基础分析流程,终于进入了正题,开始为了找寻癌细胞的转移机制而做相应的分析。首先要把关注点集中在癌细胞上,这里就体现了一开始把样本类型经行区分的好处了。在通过算法分析得到的癌细胞中(n = 1,029) ,作者发现其中混入了一部分从癌周组织提取的细胞(n = 62) 。
这些细胞源于癌周组织,但是却有着癌细胞的特征,被归类到癌细胞之中。由于在实验过程中,癌周细胞是最先被取出的样本,所以这些细胞不是源于癌细胞的污染,而是确确实实的位于癌周的癌细胞。
由此可以推断出胶质瘤的扩散机制:癌细胞从癌症内部转移至癌周组织,并且作为浸润性细胞定居在癌周组织中。
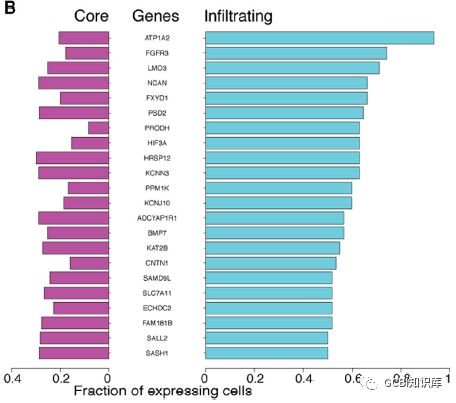
接下来,为了更好地理解浸润性细胞的分子特征,作者用DESeq2对浸润性细胞和癌症内部细胞进行差异分析。得到了1,000个左右的下调基因和250左右的上调基因。进一步筛选后,挑选出22个在浸润性细胞中高表达的基因。在这些高表达的基因中,有一些基因拥有与侵袭相关的功能。例如ATP1A2,有着能够调节细胞大小的功能;FGFR3激活细胞存活相关通路等。
而后,作者进一步通过GO数据库对细胞功能进行更专业、全面的分析。发现浸润性细胞在GO:Biological Processes层面上调富集的功能与癌细胞转移高度相关。例如细胞间粘连、阴离子运输、神经系统发育和一系列代谢过程。这些结果都已经在其它文章中被发现过。
6 .至此,作者在本次实验中,对于RNA层面的单细胞测序分析已经完成。通过单细胞RNA-seq的测序技术,作者对癌细胞、癌周细胞的特性进行了分析,并用其它文献的数据对自己的结果进行了验证。
在一些必要基础的分析过后,作者为了得到自己的目的,对癌细胞着重分析,从一千多个癌细胞中找到了自己想要的几十个浸润性细胞。这一点常规的RNA-seq完全无法做到,只有通过单细胞RNA-seq才有可能。
总之,运用适合的实验手段、合理的分析流程,才能得到了自己想要的结果。这两者必不可少,都是做好实验、写好文章的必备条件。
Reference:https://www.cell.com/cell-reports/fulltext/S2211-1247(17)31462-6?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS2211124717314626%3Fshowall%3Dtrue
本文由 GCBI学院 作者:其明技术专家 发表,转载请注明来源!